Discovering breakthroughs in fibrosis and neurodegeneration
Targeting Calpains in Fibrosis
Dimeric calpains (CAPNs 1, 2, 9) are calcium-activated cysteine proteases that regulate cellular function. Overactivation of calpains promotes signaling processes foundational to cell/tissue damage responses that eventually lead to fibrosis.
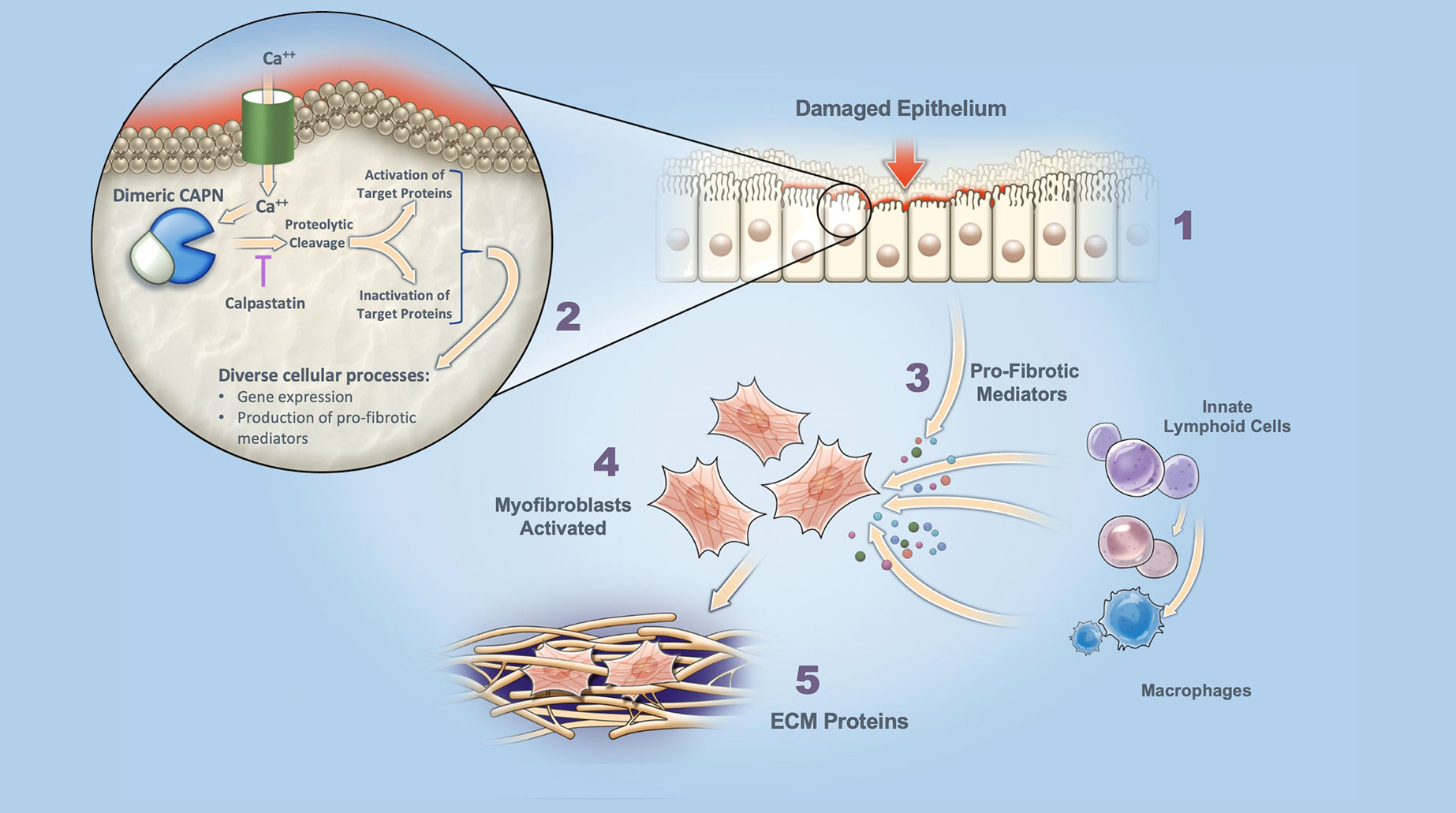
Role of Calpains in Lung Fibrosis

Dysregulated Damage Response Fibrosis is triggered by dysregulated cell / tissue damage response following epithelial injury.

Calpain (CAPN) Activation Increased intracellular calcium (Ca++) leads to activation of dimeric calpains (CAPN), which are tightly controlled by an endogenous inhibitor, calpastatin.

Release of Pro-Fibrotic Mediators Activated calpains cleave target proteins to either activate or inactivate. Excessive calpain activation (e.g., excess Ca++ influx or loss of calpastatin control) leads to elevated gene expression, and production of pro-fibrotic mediators.

Myofibroblast Activation Pro-fibrotic mediators released by damaged epithelium and associated inflammatory cells leads to myofibroblast activation / differentiation.

Secretion of ECM Proteins Activated myofibroblasts secrete extracellular matrix (ECM) proteins (scarring) that disrupt normal organ architecture and function.
Mechanism of calpain-targeted inhibition
Dimeric calpains are intracellular cysteine proteases that, when activated, selectively cleave other signaling proteins to convert them to either an active or inactive state. Consequently, normal calpain protease activity is employed as part of numerous intracellular signaling pathways. Of particular interest, calpain activity is required for differentiation/activation of myofibroblast cells that lay down extracellular collagen matrix protein (ECM, or scarring) that disrupts normal organ function seen in fibrosis.
An endogenous dimeric calpain inhibitor (calpastatin) exists within cells to counterbalance calpain activity and quickly return/maintain the system in equilibrium following calpain activation. In the diseased state, calpastatin levels can become inadequate and dimeric calpains remain “turned on” (overactivated). Persistent overactivation contributes to tissue damage and pro-inflammatory responses and underlies key steps in the fibrosis process (see graphic below).
Calpain Activity is Implicated in Tissue Damage Response AND FIBROSIS
In response to epithelial cell/tissue injury, dimeric calpain become activated and elicit a signaling response that includes release of calprotectin, a damage associated molecular pattern (DAMP) protein and expression and release of multiple pro-inflammatory/pro-fibrotic cytokines. Elevated calprotectin levels often correlate with fibrotic disease severity and progression.
In preclinical models of acute lung injury and fibrosis, calpain inhibition has been demonstrated to downregulate production of calprotectin. Inhibition of calpains has also been shown to interrupt the early steps of myofibroblast activation and differentiation, thereby attenuating the production of extracellular matrix proteins (e.g. collagen) central to the fibrosis process.
Role of Calpains in Autophagy and Neurodegeneration
Dimeric calpains are implicated as key mediators in the progression of number of neurodegenerative diseases. Notable are the autosomal-dominant (genetic) neurodegenerative diseases caused by toxic protein aggregates due to expanded poly-Q repeats, in particular Huntington’s and Machado-Joseph diseases. Dimeric calpains were shown to cleave these mutant poly-Q proteins, a necessary step leading formation of neuro-toxic protein aggregates. Furthermore, dimeric calpains are known to be key regulators (inhibitors) of cellular autophagy, the intracellular mechanism by which damaged/aggregated proteins are normally cleared to maintain normal cellular function.
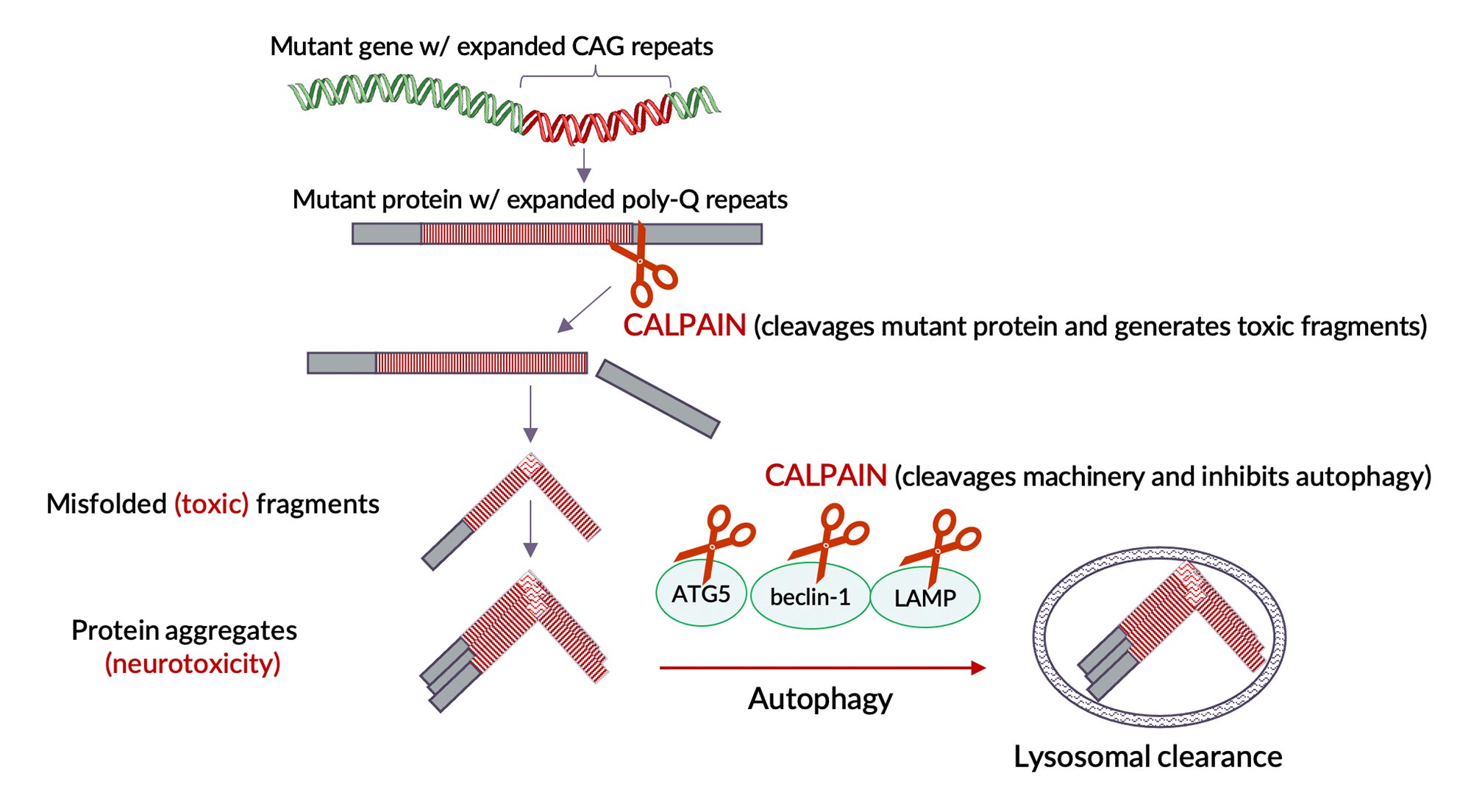
By inhibiting dimeric calpain activity we expect to reduce cleavage of mutant protein (reduce toxic protein fragments and their aggregates) and simultaneously enhance autophagy (enhance clearance of these toxic protein fragments and aggregates) that lead to neurodegeneration. Enhanced autophagy (clearance) by calpain inhibition is a general mechanism that is not limited to these orphan poly-Q neurodegenerative diseases but is applicable to a number of other neurodegenerative diseases caused by intracellular toxic proteins.
more about our science
References
Goll, 2003, Physiol Rev. 83:731.
Potz, 2016, J Nat Sci. 2(9):e218.
Blade Therapeutics in-house research
Letavernier, 2012, Cardiovasc Res. 96(1):38.
Ono, 2016, Nat Rev Drug Discov. 15(12):854.
Kim, 2019, Sci Trans Med. 11(501):2814.
Ji, 2016, Biomedical Reports 5:647.
Khanna, 2018, Ann Rheum Dis. 77(2):212.
Le, 2014, J Immunol. 193(7):3755.
Mozaffarian, 2008, J Immunol. 181(10):7243.
Pothoven, 2015, J Allergy Clin Immunol. 136(3):737.
Scaffidi, 2002, Br J Pharmacol. 136(5):793.