Discovering breakthroughs in fibrosis and neurodegeneration
OUR SCIENCE
Blade is developing potential new approaches for the treatment of debilitating fibrotic and neurodegenerative diseases
Complex, pathologic processes underlie debilitating fibrotic and neurodegenerative diseases that impact millions of people worldwide.
- FIBROSIS: The thickening or scarring of organ/tissue is characterized by the deposition of extracellular matrix proteins (fibrosis) that develops in response to aberrant cell/tissue damage. Excessive fibrosis disrupts normal organ/tissue architecture and function, and may overwhelm afflicted organs that include the liver, lung, skin, kidney, joints and eye.
- NEURODEGENERATION: Diseases where toxic protein aggregates build up within neurons and lead to cell death (neurodegeneration). Notable examples of these include autosomal dominant genetic diseases such as Huntington’s disease and Machado-Joseph disease. Both are orphan diseases caused by expanded Poly-Q repeats in the mutant proteins that form toxic protein aggregates in neurons leading to cell death.
Despite distinct etiologies and complex underlying pathophysiology, research continues to enhance our understanding of fibrotic and neurodegenerative disease processes. Today, new well-tolerated therapies that provide robust attenuation of disease progression are needed to address the high burden of these incurable conditions.
At Blade, we are experts in the novel biological pathways – including autotaxin / LPA and calpain biology – that are foundational to cell- and tissue-damage responses associated with fibrotic and neurodegenerative diseases, respectively.
Pro-fibrotic processes are stimulated by autotaxin, a key enzyme that plays a complementary role to calpains in various fibrotic diseases.
Autotaxin is a secreted enzyme that produces most of the potent phospho-lipid, lysophosphatidic acid (LPA), in the body. Autotaxin enzymatic activity converts its substrate, lysophosphatidylcholine (LPC), into LPA and choline. Excessive autotaxin levels and activity can occur in response to epithelial cell/tissue damage leading to elevated levels of LPA. LPA binds to LPA receptors on myofibroblasts, thereby triggering a signaling cascade that leads to myofibroblast activation/differentiation. View the graphic below for a step-by-step explanation.
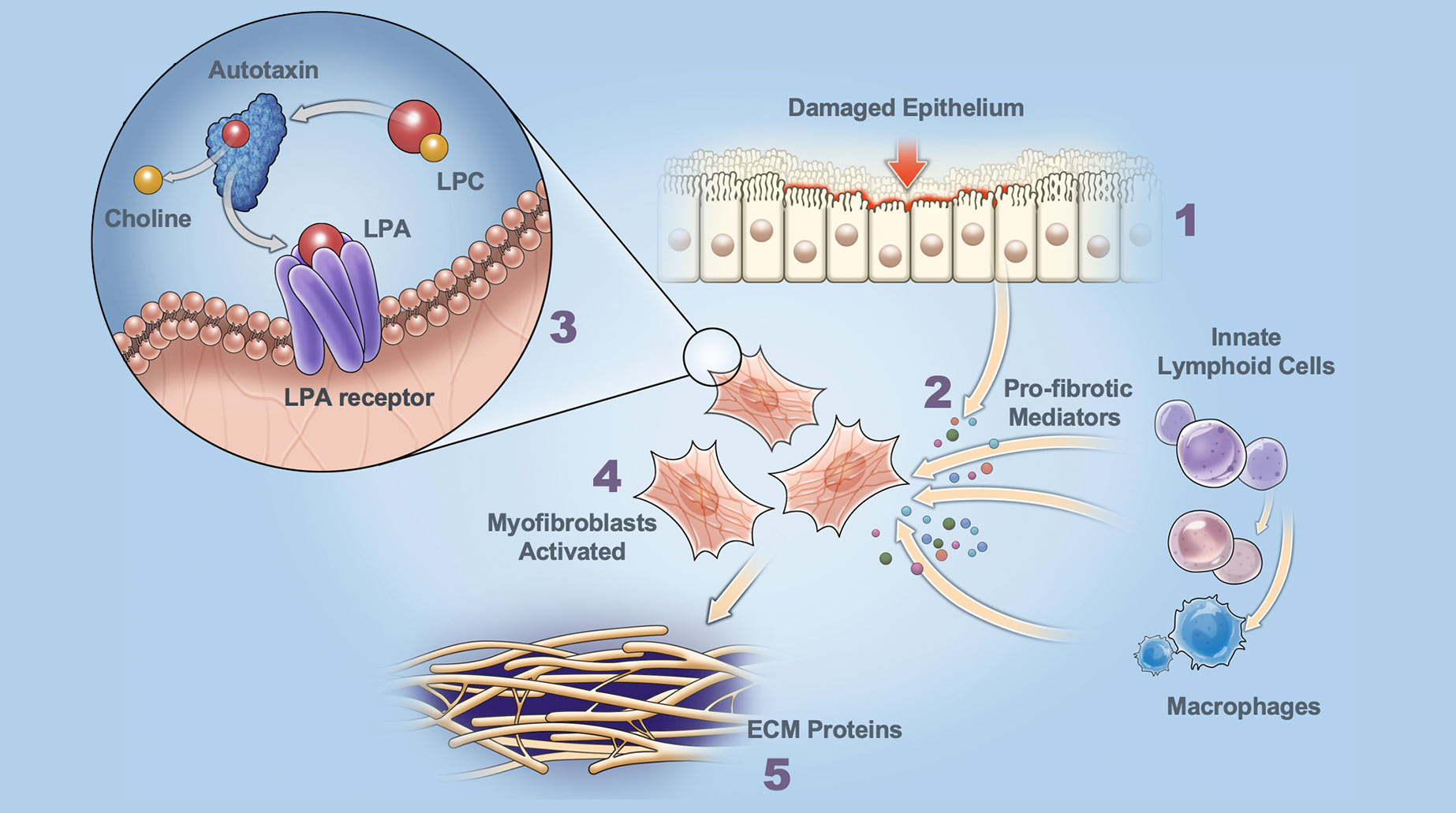
Role of Autotaxin in Fibrosis

Dysregulated Damage Response Fibrosis is triggered by dysregulated cell / tissue damage response following epithelial injury.

Release of Pro-fibrotic Mediators Pro-fibrotic mediators, cytokines and the enzyme autotaxin are released. Increased autotaxin levels produce excessive lysophosphatidic acid (LPA).

Autotaxin Production of LPA LPA binds to LPAR1 (receptor on myofibroblasts) and triggers signaling cascade resulting in migration, activation and release of additional mediators.

Myofibroblast Activation Excessive LPA activates myofibroblasts.

Secretion of ECM Proteins Activated myofibroblasts secrete ECM proteins (scarring) that disrupt normal organ architecture and function.
The autotaxin / LPA signaling cascade impacts development of disease, with aberrant LPA signaling linked to broad range of pathophysiological conditions. Increased autotaxin levels and activity are associated with liver, lung, kidney and skin fibrosis. Inhibition of the autotaxin / LPA receptor pathway has been clinically validated in idiopathic pulmonary fibrosis (IPF). In addition, autotaxin levels correlate with fibrosis severity in various liver diseases (NAFLD / NASH, viral hepatitis, ALD, cirrhosis).
Characteristics of Non-Competitive Inhibition of the Autotaxin Enzyme
The active binding site of the autotaxin enzyme offers significant potential for selective and specific inhibitors.
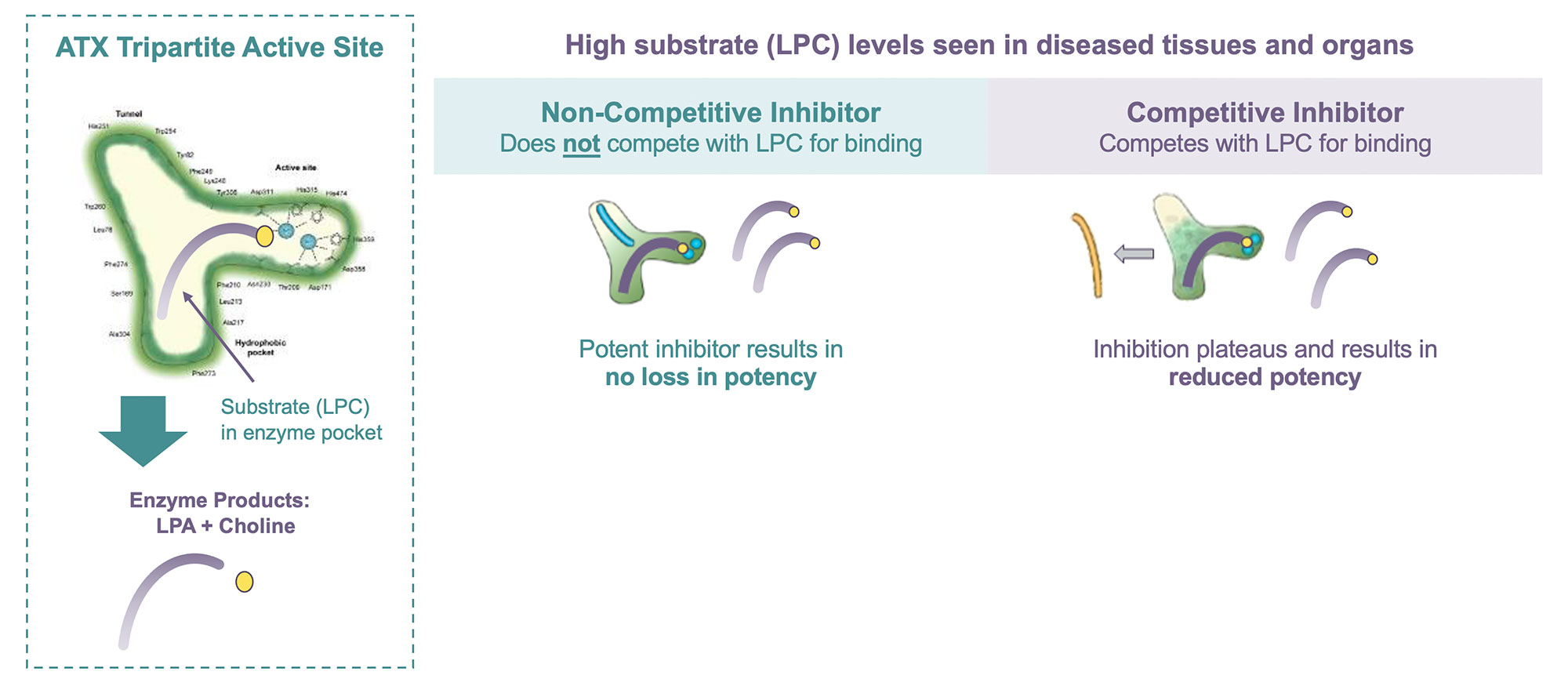
Autotaxin catalyzes the formation of LPA (lysophosphatidic acid) from LPC (lysophosphatidylcholine). LPC selectivity binds within the substrate pocket of autotaxin where it is converted into LPA plus choline and subsequently released.
Competitive inhibitors bind within the substrate binding pocket of the autotaxin active site and thereby inhibit its enzymatic activity. Since a competitive inhibitor competes with the substrate (LPC) for binding, under high substrate concentrations (as are found in fibrotic disease tissues) competitive inhibition has been shown to lose potency (reduced ability to inhibit the enzymatic activity) as it becomes displaced by high LPC levels.
Results from preclinical studies show that this is not the case with non-competitive inhibition. A non-competitive compound can inhibit the enzymatic activity whether or not the substrate (LPC) is bound and will not be displaced or lose potency under high substrate (LPC) concentrations.
More about our Science
References
Stoddard, 2015, Biomol Ther. 23(1):1.
Matralis, 2019, Med Res Rev. 39(3):976.
Maher, 2018, Lancet Resp Med. 6(8):627.
Palmer, 2018, Chest. 154(5):1061.
Galapagos company news release. 10 Sept 2020.
Perrakis, 2014 J Lipid Res. 55:1010-1018.
Biochem J. 2014 Oct 1;463(1):157-65.
The Journal of Biochemistry, Volume 148, Issue 1, July 2010, Pages 13–24.